Cellular respiration is the process of oxidizing food molecules, like glucose, to carbon dioxide and water. The energy released is trapped in the form of ATP for use by all the energy-consuming activities of the cell.
The process occurs in two phases: Mitochondria have:
Mitochondria have:
The matrix contains a complex mixture of soluble enzymes that catalyze the respiration of pyruvic acid and other small organic molecules. The enzymatic steps in the breakdown of pyruvic acid make up the citric acid cycle.

The electrons of NADH and FADH2 are transferred to the respiratory chain.
 The four complexes of integral membrane proteins that make up the respiratory chain accomplish the following:
The four complexes of integral membrane proteins that make up the respiratory chain accomplish the following:
The energy released as electrons pass down the gradient from NADH to oxygen is harnessed by the
.gif)
As their concentration increases there (which is the same as saying that the pH decreases), a strong diffusion gradient is set up. The only exit for these protons is through the ATP synthase complex. As in chloroplasts, the energy released as these electrons flow down their gradient is harnessed to the synthesis of ATP. The process is called chemiosmosis and is an example of facilitated diffusion.
One-half of the 1997 Nobel Prize in Chemistry was awarded to Paul D. Boyer and John E. Walker for their discovery of how ATP synthase works.
It depends.
It is tempting to try to view the synthesis of ATP as a simple matter of stoichiometry (the fixed ratios of reactants to products in a chemical reaction). But (with 3 exceptions) it is not.
Most of the ATP is generated by the proton gradient that develops across the inner mitochondrial membrane. The number of protons pumped out as electrons drop from NADH through the respiratory chain to oxygen is theoretically large enough to generate, as they return through ATP synthase, 3 ATPs per electron pair (but only 2 ATPs for each pair donated by FADH2). With 12 pairs of electrons removed from each glucose molecule, 10 by NAD+ (so 10x3=30); 2 by FADH2 (so 2x2=4), this could generate 34 ATPs. Add to this the 4 ATPs that are generated by the 3 exceptions and one arrives at 38.
But the energy stored in the proton gradient is used for a number of other mitochondrial functions such as the active transport of a variety of essential molecules and ions through the mitochondrial membranes. So the actual yield of ATP as mitochondria respire varies with conditions.
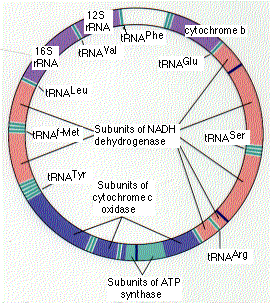
The human mitochondrion contains 5-10 identical, circular molecules of DNA. Each consists of 16,569 base pairs carrying the information for 37 genes which encode:
The rRNA and tRNA molecules are used in the machinery that synthesizes the 13 polypeptides.
The 13 polypeptides are subunits of the protein complexes in the inner mitochondrial membrane, including subunits of NADH dehydrogenase, cytochrome c oxidase, and ATP synthase. However, each of these protein complexes also requires subunits that are encoded by nuclear genes, synthesized in the cytosol, and imported from the cytosol into the mitochondrion.
Although many different organs may be affected, disorders of the brain and muscles are the most common. Perhaps this reflects the great demand for energy of both these organs.
Some of these disorders are inherited in the germline. In every case, the mutant gene is received from the mother because none of the mitochondria in sperm survives in the fertilized egg. Other disorders are somatic; that is, the mutation occurs in the somatic tissues of the individual.
Many of the features of the mitochondrial genetic system resemble those found in prokaryotes like bacteria. This has strengthened the theory that mitochondria are the evolutionary descendants of a prokaryote that established an endosymbiotic relationship with the ancestors of eukaryotic cells early in the history of life on earth. However, many of the genes needed for mitochondrial function have since moved to the nuclear genome.
The recent sequencing of the complete genome of Rickettsia prowazekii has revealed a number of genes closely related to those found in mitochondria. Perhaps rickettsias are the closest living descendants of the endosymbionts that became the mitochondria of eukaryotes.
| Further discussion of the evolutionary implications of mtDNA. |
| Welcome&Next Search |